Welcome to another exciting StatQuest adventure! In this part of our series on edgeR and DESeq2, we will delve into the concept of independent filtering. Independent filtering, also known as mitigating the multiple testing problem, is an essential step in conducting reliable statistical tests.
Contents
The Challenge of Multiple Testing
Before we dive into independent filtering, let’s briefly recap the challenge of multiple testing. Whenever we perform a statistical test, there is a chance that our conclusion could be wrong. To understand this better, check out the StatQuest on p-values in a nutshell. Essentially, when we set a significance threshold of p < 0.05, we accept that 5% of the time, we may report a false positive.
This is not a big deal when we test a small number of genes. However, when we test thousands of genes, such as in cancer cell analysis, even a 5% false positive rate can lead to a considerable number of incorrect results. Therefore, we need techniques like FDR (false discovery rate) and the Benjamini-Hochberg method to compensate for this problem. Refer to the StatQuest on FDR for more details on these methods.
The Problem of Overlapping Values
However, there is still another problem to consider. Let’s imagine we have two distributions representing the weights of two different mouse strains. In most cases, if we weigh a mouse from one strain, the value will fall within a specific range. Similarly, if we weigh a mouse from the other strain, its weight will also fall within a particular range.
However, occasionally, we may encounter overlapping weight values, resulting in a false negative. This is a case where a statistical test fails to detect a significant difference between the two distributions.
To visualize this, let’s take 1,000 samples from these distributions and perform 1,000 t-tests. In the histogram of these 1,000 p-values, we observe a group of p-values less than 0.05, representing true positives, and a small number of p-values greater than 0.05, indicating false negatives.
Independent Filtering to the Rescue
To address this problem, independent filtering comes into play. Both edgeR and DESeq2 have methods to filter out genes with low read counts, which are not informative and may contribute to false positives.
The general idea is to remove genes with very low read counts from the dataset. Even though some of these genes may be biologically interesting, it is challenging to obtain accurate read counts if there are only a few transcripts in one sample type compared to another.
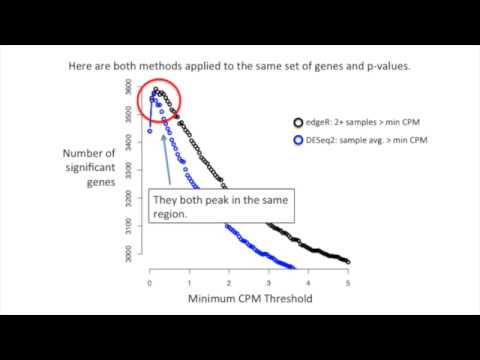
EdgeR recommends removing all genes with less than one count per million (CPM) in at least two samples. CPM compensates for differences in read depth between libraries. For example, if a dataset has over 5 million total reads, the CPM scaling factor is 5.32. Genes with CPM values greater than 1 in two or more samples are retained, while others are filtered out.
DESeq2 takes a slightly different approach. It calculates the average normalized read counts across all samples. Genes with average CPM values greater than the minimum CPM threshold are kept. DESeq2 then fits a curve to the significant genes versus quantiles and determines the threshold as the maximum location on the curve minus the standard deviation.
It’s important to note that while both methods achieve similar results, DESeq2’s approach is more flexible, allowing the use of quantiles as the x-axis to define the cutoff.
Recommendations for Filtering Genes
When using edgeR, it’s recommended to adjust the CPM cutoff after calculating the p-values. Explore different cutoff values to achieve the optimal gene selection criteria for your dataset. To simplify this process, a template code is available on my website. Remember to cite both edgeR and DESeq2 if you choose to combine their methods.
If you decide to use DESeq2, be cautious about outliers when there are only two samples per category. DESeq2 offers an outlier detection method to address this issue, which we will cover in a future StatQuest.
In conclusion, independent filtering plays a crucial role in mitigating the multiple testing problem. Both edgeR and DESeq2 provide methods to filter out genes with low read counts, improving the reliability of statistical tests. By carefully selecting the CPM cutoff and considering outliers, you can enhance the accuracy of your analysis.
Thank you for joining us on this StatQuest adventure! Stay tuned for more exciting explorations in the world of statistics. Until then, keep questing on!
FAQs
Q: Can you recommend specific CPM cutoff values for my dataset?
A: CPM cutoff values depend on the specific characteristics of your dataset, such as sequencing depth and biological context. It’s best to choose cutoffs based on your domain knowledge and experimental design. You can also experiment with different cutoff values to observe the impact on the number of significant genes.
Q: How can I determine if my data requires additional outlier detection methods?
A: If you are using DESeq2 and have more than two samples per category, the built-in outlier detection method will automatically kick in. However, for datasets with only two samples per category, you may need to consider additional outlier detection techniques. Consult the relevant literature or seek advice from experts in your field for guidance.
Q: Can I use both edgeR and DESeq2 methods together?
A: Yes, it is possible to combine the filtering methods of edgeR and DESeq2. You can first apply edgeR’s CPM cutoff, and then utilize DESeq2’s approach to find the optimal CPM threshold. However, ensure that you cite both publications to give proper credit to each method.
Conclusion
Independent filtering is a crucial step in mitigating the multiple testing problem. Both edgeR and DESeq2 provide methods to filter out genes with low read counts, enhancing the accuracy of statistical tests. By carefully selecting appropriate cutoffs and considering outliers, you can improve the reliability of your gene expression analysis. Keep exploring and quest on! Techal
