Welcome to Techal, where we dive deep into the world of technology! Today, we’re going to explore DESeq2, a powerful program used for identifying differential gene expression. Specifically, we’ll focus on the first part of DESeq2: library normalization.
Contents
Understanding Library Normalization
Library normalization is the process of adjusting for differences in library sizes when comparing gene expression. This is crucial because different samples might have varying numbers of reads mapped to them, which can skew the results.
To illustrate this concept, let’s assume we have two samples with different numbers of reads mapped to them. Sample 1 has 635 reads, while sample 2 has 1270 reads. In this scenario, the read counts for each gene in sample 2 are twice that of sample 1, not due to biology, but because of sequencing depth.
Library normalization tackles two main problems:
Adjusting for Differences in Library Sizes
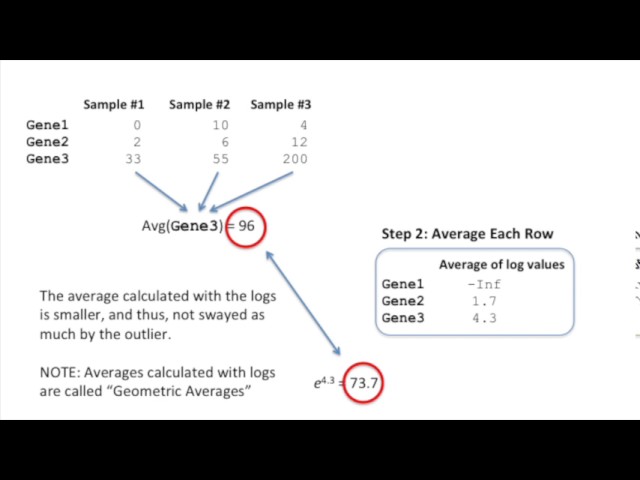
The first problem library normalization addresses is adjusting for differences in library sizes. To achieve this, DESeq2 introduces a scaling factor for each sample. This factor takes into account both the read depth and the composition of the library. By taking the logarithm of the read counts and averaging them, DESeq2 ensures that outliers don’t heavily influence the scaling factor calculation.
Adjusting for Differences in Library Composition
The second problem library normalization addresses is adjusting for differences in library composition. In high-throughput sequencing, comparing gene expression between different tissue types or when gene transcription is affected by factors like knockout experiments can lead to differences in the composition of the library. These differences can impact the expression levels of specific genes.
The DESeq2 Normalization Process
To better understand how DESeq2 normalizes read counts, let’s break down the process into steps:
- Take the logarithm of all the read counts.
- Average the log values for each gene, filtering out genes with an average log value of infinity.
- Subtract the average log value from each log count, obtaining the log ratios of each gene compared to the average.
- Calculate the median of the log ratios for each sample, providing a more balanced measurement.
- Convert the medians back to normal numbers using the exponential function of the programming language R, which DESeq2 is built upon.
- Use the scaling factors obtained in the previous step to divide the original read counts, effectively normalizing the data.
- Obtain the final scaled read counts that are ready for further analysis.
Conclusion
DESeq2’s library normalization process addresses the challenges of comparing gene expression data with different library sizes and compositions. By taking logarithms, averaging, and utilizing scaling factors, DESeq2 ensures that the analysis focuses on genes expressed at similar levels across all samples. This normalization step sets the foundation for accurate identification of differentially expressed genes.
Stay tuned for our next article, where we’ll explore how DESeq2’s counterpart, EdgeR, normalizes read counts. For more insightful analysis and comprehensive guides on all things technology, visit Techal.
FAQs
Q: Why is library normalization important in gene expression analysis?
A: Library normalization is crucial in gene expression analysis as it adjusts for differences in library sizes and compositions, ensuring accurate comparisons between samples.
Q: How does DESeq2 handle differences in library sizes and compositions during normalization?
A: DESeq2 uses a combination of logarithmic transformations, averaging, and scaling factors to address differences in library sizes and compositions, thereby normalizing the read counts.
Q: What does the scaling factor in DESeq2 represent?
A: The scaling factor in DESeq2 represents the adjustment needed to account for differences in library sizes and compositions, enabling fair comparisons between samples.
Q: Why does DESeq2 use logarithmic transformations in the normalization process?
A: DESeq2 uses logarithmic transformations to stabilize the read counts and prevent outliers from skewing the normalization results. The logarithms also enable easier comparison and analysis of gene expression levels.
Q: Can DESeq2 be used with various types of data sets?
A: Yes, DESeq2 is designed to handle diverse data sets, making it a versatile tool for gene expression analysis across different experimental conditions and sample types.
Q: Are there any specific genes that DESeq2 focuses on during normalization?
A: DESeq2 aims to focus on housekeeping genes, which are genes that are transcribed at similar levels regardless of tissue type or experimental conditions. These genes serve as reference points for normalization.
References
- DESeq2: https://techal.org/
